| Step 1: An acid/base reaction. Since we only have a weak nucleophile and apoor electrophile we need to activate the amide. Protonation of the amidecarbonyl makes it more electrophilic. | 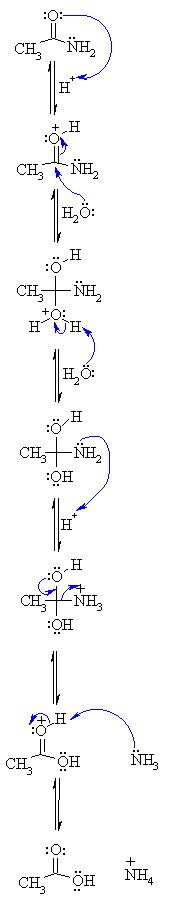 |
| Step 2: The water O functions as the nucleophile attacking the electrophilicCin the C=O, with the electrons moving towards the oxonium ion, creatingthe tetrahedral intermediate. | |
| Step 3: An acid/base reaction. Deprotonate the oxygen that came from the watermolecule to neutralise the charge. | |
| Step 4: An acid/base reaction. Need to make the -NH2leave, but need to convert it into a good leaving group first byprotonation. | |
| Step 5: Use the electrons of an adjacent oxygen to help "push out" the leavinggroup, a neutral ammonia molecule. | |
| Step 6: An acid/base reaction. Deprotonation of the oxonium ion reveals thecarbonyl in the carboxylic acid product and regenerates the acid catalyst. |
Thursday, May 19, 2011
MECHANISM OF THE ACID CATALYSED HYDROLYSIS OF AMIDES
REACTION OF RMgX WITH AN NITRILE
| Step 1: The nucleophilic C in the organometallic reagent adds to the electrophilic C in the polar nitrile group. Electrons from the CºN move to the electronegative N creating an intermediate imine salt complex. |   |
| Step 2: An acid/base reaction. On addition of aqueous acid, the intermediate salt protonates giving the imine. | |
| Step 3: An acid/base reaction. Imines undergo nucleophilic addition, but require activation by protonation (i.e. acid catalysis) | |
| Step 4: Now the nucleophilic O of a water molecule attacks the electrophilic C with the p bond breaking to neutralize the change on the N. | |
| Step 5: An acid/base reaction. Deprotonate the O from the water molecule to neutralize the positive charge. | |
| Step 6: An acid/base reaction. Before the N system leaves, it needs to be made into a better leaving group by protonation. | |
| Step 7: Use the electrons on the O in order to push out the N leaving group, a neutral molecule of ammonia. | |
| Step 8: An acid/base reaction. Deprotonation reveals the carbonyl group of the ketone product. | |
MECHANISM OF THE ACID catalyzed HYDROLYSIS OF NITRILES

HYDROLYSING NITRILES
The hydrolysis of nitriles
Introduction
When nitriles are hydrolysed you can think of them reacting with water in two stages - first to produce an amide, and then the ammonium salt of a carboxylic acid.
For example, ethanenitrile would end up as ammonium ethanoate going via ethanamide.
![]()
![]()
In practice, the reaction between nitriles and water would be so slow as to be completely negligible. The nitrile is instead heated with either a dilute acid such as dilute hydrochloric acid, or with an alkali such as sodium hydroxide solution.
The end result is similar in all the cases, but the exact nature of the final product varies depending on the conditions you use for the reaction.
Acidic hydrolysis of nitriles
The nitrile is heated under reflux with dilute hydrochloric acid. Instead of getting an ammonium salt as you would do if the reaction only involved water, you produce the free carboxylic acid.
For example, with ethanenitrile and hydrochloric acid you would get ethanoic acid and ammonium chloride.
![]()
![]()
Why is the free acid formed rather than the ammonium salt? The ethanoate ions in the ammonium ethanoate react with hydrogen ions from the hydrochloric acid to produce ethanoic acid. Ethanoic acid is only a weak acid and so once it has got the hydrogen ion, it tends to hang on to it.
Alkaline hydrolysis of nitriles
The nitrile is heated under reflux with sodium hydroxide solution. This time, instead of getting an ammonium salt as you would do if the reaction only involved water, you get the sodium salt. Ammonia gas is given off as well.
For example, with ethanenitrile and sodium hydroxide solution you would get sodium ethanoate and ammonia.
![]()
![]()
The ammonia is formed from reaction between ammonium ions and hydroxide ions.
If you wanted the free carboxylic acid in this case, you would have to acidify the final solution with a strong acid such as dilute hydrochloric acid or dilute sulphuric acid. The ethanoate ion in the sodium ethanoate will react with hydrogen ions as mentioned above.
Structure of P4O10
Many inorganic solids have a 3-dimensional structure of the complex. Illustration different from the same compound will help us understand the structure. In the case of complex inorganic compounds, depicts the bond between atoms, such as those used in organic compounds usually cause confusion. Anion in most oxides, sulfides or metal halide to form tetrahedral or octahedral cations surrounding the metal. Although there are no bonds between the anions, the structure will be simplified if the structure is illustrated with anion polyhedra sharing corners, sides or front. In illustration of this kind, the metal atoms are usually ignored.
As mentioned ionic structures can be regarded as a composition terjejal anions. Figure 2.12 illustrates this representation for the three molecular phosphorus pentoxide P2O5 (= P4O10)

As mentioned ionic structures can be regarded as a composition terjejal anions. Figure 2.12 illustrates this representation for the three molecular phosphorus pentoxide P2O5 (= P4O10)
Hydrolysis of carboxylic acid derivatives (8)
watch this video!!!
this video talking about Base-catalyzed amide hydrolysis.....
Hydrolysis of carboxylic acid derivatives (7)
This video talking about Acid-catalyzed amide hydrolysis.........
Saturday, May 7, 2011
Carboxylic acid derivatives
The carboxyl group (abbreviated -CO2H or -COOH) is one of the most widely occurring functional groups in chemistry as well as biochemistry. The carboxyl group of a large family of related compounds called Acyl compounds or Carboxylic Acid Derivatives.
All the reactions and compounds covered in this section will yield Carboxylic Acids on hydrolysis, and thus are known as Carboxylic Acid Derivatives. Hydrolysis is one example of Nucleophilic Acyl Substitution, which is a very important two step mechanism that is common in all reactions that will be covered here.
The systematic IUPAC nomenclature for carboxylic acid derivatives is different for the various compounds which are in this vast category, but each is based upon the name of the carboxylic acid closest to the derivative in structure. Each type is discussed individually below.
Acyl Groups
Acyl groups are named by stripping the -ic acid of the corresponding carboxylic acid and replacing it with -yl.
EXAMPLE:
CH3COOH = acetic acid
CH3COO-R = acetyl-R
Acyl Halides
Simply add the name of the attached halide to the end of the acyl group.
EXAMPLE:
CH3COOH = acetic acid
CH3COBr = acetyl bromide
Carboxylic Acid Anhydrides
A carboxylic acid anhydride ([RC=O]O[O=CR]) is a carboxylic acid (COOH) that has an acyl group (RC=O) attached to its oxygen instead of a hydrogen. If both acyl groups are the same, then it is simply the name of the carboxylic acid with the word acid replaced with anhydride. If the acyl groups are different, then they are named in alphabetical order in the same way, with anhydride replacing acid.
EXAMPLE:
CH3COOH = acetic acid
CH3CO-O-OCCH3 = Ethanoic Anhydride
Esters
Esters are created when the hydrogen on a carboxylic acid is replaced by an alkyl group. Esters are known for their pleseant, fruity smell and taste, and they are often found in both natural and artificial flavors. Esters (RCOOR1) are named as alkyl alkanoates. The alkyl group directly attached to the oxygen is named first, followed by the acyl group, with -ate replacing -yl of the acyl group.
EXAMPLE:
CH3COOH = acetic acid
CH3COOCH2CH2CH2CH3 = acetyl butanoate
Amides
Amides which have an amino group (-NH2) attached to a carbonyl group (RC=O) are named by replacing the -oic acid or -ic acid of the corresponding carboxylic acid with -amide.
EXAMPLE:
CH3COOH = acetic acid
CH3CONH2 = acetamide
Nitriles
Nitriles (RCN) can be viewed a nitrogen analogue of a carbonyl and are known for their strong electron withdrawing nature and toxicity. Nitriles are named by adding the suffix -nitrile to the longest hydrocarbon chain (including the carbon of the cyano group). It can also be named by replacing the -ic acid or -oic acid of their corresponding carboxylic acids with -onitrile. Functional class IUPAC nomenclature may also be used in the form of alkyl cyanides.
EXAMPLE:
CH3CH2CH2CH2CN = butonitrile or butyl cyanide
Structure and Reactivity
Stability and reactivity have an inverse relationship, which means that the more stable a compound, generally the less reactive - and vice versa. Since acyl halides are the least stable group listed above, it makes sense that they can be chemically changed to the other types. Since the amides are the most stable type listed above, it should logically follow that they cannot be easily changed into the other molecule types, and this is indeed the case.
The stability of any type of carboxylic acid derivative is generally determined by the ability of its functional group to donate electrons to the rest of the molecule. In essence, the more electronegative the atom or group attached to carbonyl group, the less stable the molecule. This readily explains the fact that the acyl halides are the most reactive, because halides are generally quite electronegative. It also explains why acid anhydrides are unstable; with two carbonyl groups so close together the oxygen in between them cannot stabilize both by resonance - it can't loan electrons to both carbonyls.
The following derivative types are ordered in decreasing reactivity (the first is the most reactive):
Acyl Halides (CO-X) > Acyl Anhydrides (-CO-O-OCR) > Acyl Thioester (-CO-SR) > Acyl Esters (-CO-OR) > Acyl Amides (-CO-NR2)
As mentioned before, any substance in the preceding list can be readily transformed into a substance to its right; that is, the more reactive derivative types (acyl halides) can be directly transformed into less reactive derivative types (esters and amides). Every type can be made directly from carboxylic acid (hence the name of this subsection) but carboxylic acid can also be made from any of these types.
Reactions of Carboxylic Acids and Their Derivatives
Carboxylic Acids
1) As acids:
RCO2H + NaOH ----> RCO2-Na+ + H2O RCO2H + NaHCO3 ----> RCO2-Na+ + H2O + CO2
2) Reduction:
RCO2H + LiAlH4 --- (1) Et2O -- (2) H2O ----> RCH2OH
3) Conversion to acyl chlorides:
RCO2H -----SOCl2 or PCl5 ----> RCOCl
4) Conversion to esters (Fischer esterfication):
RCOOH + R'-OH <--- HA ---> RCOOR' + H2O
5) Conversion to amides:
RCO2H -----SOCl2 or PCl5 ----> RCOCl + NH3 <------> RCOO-NH4+ --- heat ---> R-CONH2 + H2O
6) Decarboxylation: (Note: you need a doubly-bonded oxygen (carbonyl) two carbons away for this reaction to work)
RCOCH2COOH --- heat ---> R-COCH3 + CO2 HOCOCH2COOH --- heat ---> CH3COOH + CO2
Acyl Chlorides
1) Conversion to acids:
R-COCl + H2O ----> R-COOH + HCl
2) Conversion to anhydrides:
R-COCl + R'COO- ----> R-CO-O-COR' + Cl-
3) Conversion to esters:
R-COCl + R'-OH --- pyridine ---> R-COOR' + Cl- + pyr-H+
4) Conversion to amides:
R-COCl + R'NHR" (excess) ----> R-CONR'R" + R'NH2R"Cl
R' and/or R" may be H
5) Conversion to ketones:
Friedel-Crafts acylation
R-COCl + C6H6 --- AlCl3 ---> C6H5-COR
Reaction of Dialkylcuprates (also known as a Gilman reagent)
R-COCl + R'2CuLi ----> R-CO-R'
6) Conversion to aldehydes:
R-COCl + LiAlH[OC(CH3)3]3 --- (1) Et2O (2) H2O ---> R-CHO
Acid Anhydrides
1) Conversion to acids:
(R-CO)2-O + H2O ----> 2 R-COOH
2) Conversion to esters:
(R-CO)2-O + R'OH ----> R-COOR' + R-COOH
3) Conversion to amides:
(R-CO)2-O + H-N-(R'R") ----> R-CON-(R'R") + R-COOH
R' and/or R" may be H.
4) Conversion to aryl ketones (Friedel-Crafts acylation):
(R-CO)2-O + C6H6 --- AlCl3 C6H5-COR + R-COOH
Esters
1) Hydrolysis:
R-COOR' + H2O <--- HA ---> R-COOH + R'-OH R-COOR' + OH- ----> RCOO- + R'-OH
2) Transesterification (conversion to other esters):
R-COOR' + R"-OH <--- HA ---> R-COO-R" + R'-OH
3) Conversion to amides:
R-COOR' + HN-(R"R"') ----> R-CON-(R"R"') + R'-OH
R" and/or R"' may be H
4) Reaction with Grignard reagents:
R-COOR' + 2 R"MgX --- Et2O ---> R-C-R"2OMgX + R'OMgX ---> H3O+ R-C-R"2OH
The intermediate and final product is a tetrahedral carbon with two R" attached directly to the carbon along with R and OH/OMgX
X = halogen.
5) Reduction:
R-COOR' + LiAlH4 --- (1) Et2O (2) H2O ---> R-CH2OH + R'-OH
Amides
1) Hydrolysis:
R-CON(R'R") + H3O+ --- H2O ---> R-COOH + R'-N+H2R" R-CON(R'R") + OH- --- H2O ---> R-COO- + R'-NHR"
R,R' and/or R" may be H.
2) Dehydration (conversion to nitriles):
R-CONH2 --- P4O10, heat, (-H2O) ---> R-CN
[edit] Nitriles
1) Hydrolysis:
R-CN --- H3O+,heat ---> RCOOH R-CN --- OH-,H2O,heat ---> RCOO-
2) Reduction to aldehyde:
R-CN --- (1) (i-Bu)2AlH (2) H2O ---> R-COH
(i-Bu)2AlH = DIBAL-H
3) Conversion to ketone (by Grignard or organolithium reagents):
R-CN + R"-M --- (1) Et2O (2) H3O+ ---> R-COR"
M = MgBr (Grignard reagent) or Li (organolithium reagent)
Mechanisms
A common motif in reactions dealing with carboxylic acid derivatives is the tetrahedral intermediate. The carbonyl group is highly polar, with the carbon having a low electron density, and the oxygen having a high electron density. With an acid catalyst, a H+ is added to the oxygen of the carbonyl group, increasing the positive charge at the carbon atom. A nucleophile can then attack the carbonyl, creating a tetrahedral intermediate.
For example, in Fischer esterification, the mechanism can be outlined thus: 1) H+ is added to carbonyl oxygen 2) Oxygen atom of the alcohol adds to the carbonyl carbon 3) Proton transfer from alcohol oxygen to carboxyl oxygen 4) Water molecule ejected from tetrahedral intermediate, double bond forms, recreating the carbonyl 5) H+ is removed from carbonyl oxygen
Homo-Lumo Interactions
A fundamental principle: all steps of all heterolytic reaction mechanisms are either Bronsted or Lewis acid-base reactions
- They involve either proton transfer (Bronsted), or unshared pair/empty orbital interactions (Lewis).
- When the interacting atomic orbitals are considered, the Bronsted reactions can be seen as simply a special case of the Lewis, in which the empty orbital is the antibonding orbital of the H-X bond.
In short, all heterolytic reactions are just examples of interactions between filled atomic or molecular orbitals and empty atomic or molecular orbitals - that is, Lewis acid-base reactions. Here is a diagram to explain this point:

The interaction of any two atomic or molecular orbitals, as you learned in general chemistry, produces two new orbitals.
- One of the new orbitals is higher in energy than the original ones (the antibonding orbital), and one is lower (the bonding orbital).
- When one of the initial orbitals is filled with a pair of electrons (a Lewis base), and the other is empty (a Lewis acid), we can place the two electrons into the lower energy of the two new orbitals.
- The "filled-empty" interaction therefore is stabilizing.
When we are dealing with interacting molecular orbitals, the two that interact are generally
- The highest energy occupied molecular orbital (HOMO) of one molecule,
- The lowest energy unoccupied molecular orbital (LUMO) of the other molecule.
- These orbitals are the pair that lie closest in energy of any pair of orbitals in the two molecules, which allows them to interact most strongly.
- These orbitals are sometimes called the frontier orbitals, because they lie at the outermost boundaries of the electrons of the molecules.
Here is the filled-empty interaction redrawn as a HOMO-LUMO interaction.

Let's look at some examples. First, a reaction that you would have categorized as a Lewis acid-base reaction when you were studying general chemistry:
NH3 has an unshared pair on nitrogen, occupying the HOMO (it is generally true that unshared pairs occupy HOMOs). BH3 has an empty valence orbital on B, since B is a Group II element. This is the LUMO.
Here are pictures of the two orbitals from AM1 semi-empirical molecular orbital calculations:
| NH3 HOMO | BH3 LUMO |
|---|---|
 |  |
The HOMO-LUMO energy diagram above describes the formation of a bond between N and B.
Now let's try a slightly more complex case. Here's a typical Bronsted acid-base reaction:

The curly arrows track which bonds are made, and which are broken, but they do not indicate what orbitals are involved.
- Water is both a Bronsted base (capable of accepting a proton) and a Lewis base, with one of its unshared pairs (the HOMO).
- H-Cl is a Bronsted acid, capable of donating a proton, but it also is a Lewis acid, using the s* orbital of the H-Cl bond (the LUMO).
- Here are pictures of the relevant HOMO and LUMO, again from AM1 semi-empirical molecular orbital calculations:
H2O HOMO HCl LUMO 

- The interaction stabilizes the unshared pair of the oxygen, while simultaneously breaking the H-Cl bond because the interaction is with the antibonding orbital.
Here are the relevant orbitals:
| OH- HOMO | CH3-Cl LUMO |
|---|---|
 |  |
The interaction stabilizes the unshared pair of the oxygen, while simultaneously breaking the CH3-Cl bond because the interaction is with the antibonding orbital.
Other examples include the reaction of alkenes with H-X, where the HOMO is the p MO of the alkene and the LUMO is the H-X s* orbital:

and the capture of the carobcation in an SN1 reaction by nucleophile:

You should need no reminder that the carbocation is stabilized by a filled-empty interaction between the empty p orbital of the positive carbon and the s orbital of an adjacent C-H or C-C bond
In short, all heterolytic reactions proceed because the energy of a pair of electrons is lowered by the interaction of a filled atomic or molecular orbital with an empty one.
The same reasoning can be appllied to bimolecular pericyclic reactions like the Diels-Alder cycloaddition.
Subscribe to:
Posts (Atom)